Popis Costellovho syndrómu. Prípad zriedkavej choroby u novorodenca. Detská republikánska klinická nemocnica Ministerstva zdravotníctva Tatarskej republiky
Costellov syndróm tiež nazývaný faciocutaneoskeletálny syndróm alebo fCS syndróm , je zriedkavá genetická porucha, ktorá postihuje mnohé časti tela. Vyznačuje sa spomaleným vývojom a duševnými schopnosťami, výraznými črtami tváre, neobvykle pružnými kĺbmi a voľnými záhybmi kože navyše, najmä na rukách a nohách. Srdcové abnormality sú časté, vrátane veľmi rýchleho srdcového rytmu (tachykardia), štrukturálnych chýb srdca a zväčšenia srdcového svalu (hypertrofická kardiomyopatia). Deti s Costellovým syndrómom môžu byť po narodení veľké, ale rastú pomalšie ako iné deti a majú výživové ťažkosti. Neskôr v živote majú ľudia s týmto ochorením relatívne nízku postavu a veľa znížených hladín rastových hormónov. Toto je RASopatia.
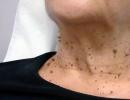
Od raného detstva majú ľudia s Costellovým syndrómom zvýšené riziko vzniku určitých rakovinových a nerakovinových nádorov. Najčastejšie sa vyskytujú malé výrastky nazývané papilómy benígne nádory videný s týmto stavom. Zvyčajne sa vyvíjajú okolo alebo v blízkosti nosa a úst konečník... Najbežnejšou rakovinou spojenou s Costellovým syndrómom je nádor mäkkých tkanív nazývaný rabdomyosarkóm. U detí a dospievajúcich s týmto ochorením boli tiež hlásené ďalšie druhy rakoviny, vrátane nádoru, ktorý sa vyskytuje vo vývoji nervových buniek (neuroblastóm) a formy rakoviny močového mechúra (prechodný bunkový karcinóm).
Jackov Costello, novozélandský pediatr v roku 1977, objavil Costelloov syndróm a prvé správy o tomto syndróme sú zaznamenané v Australian Pediatric Journal, zväzok 13, č. 2 v roku 1977.
príznaky a symptómy
genetika
Costellov syndróm je spôsobený ktoroukoľvek z najmenej piatich rôznych mutácií v HRAS gén na chromozóme 11. Tento gén obsahuje pokyny na výrobu proteínu H-Ras, ktorý pomáha riadiť rast a delenie buniek. Mutácie, ktoré spôsobujú Costellov syndróm, vedú k produkcii proteínu H-Ras, ktorý je neustále aktívny. Namiesto spustenia bunkového rastu v reakcii na určité signály z vonkajšej strany bunky, hyperaktívny proteín smeruje bunku k ďalšiemu rastu a deleniu. Toto zlyhanie pri zastavení bunkového delenia môže predisponovať postihnutých k vývoju benígnych a malígnych nádorov. Zostáva nejasné, ako mutácie prebiehajú HRAS spôsobiť ďalšie príznaky Costellovho syndrómu, ale veľa znakov a symptómov môže byť výsledkom bunkovej proliferácie a abnormálneho delenia buniek.
Klinická diagnóza zriedkavého dedičného ochorenia - Costellov syndróm
T.N. Vasina, T.I. Zubtsova, S.N. Stavtseva, T.A. Kirsanova
Klinická diagnóza zriedkavého dedičného ochorenia: Costellov syndróm
T.N. Vasina, T.I. Zubtsova, S.N. Stavtseva, T.A. Kirsanova
Oryol Medical Institute
Uvádzame literárne údaje a vlastné klinické pozorovania dieťaťa s Costellovým syndrómom. Mimoriadne zriedkavé genetický syndróm charakterizovaná pretrvávajúcou dysfágiou vyžadujúcou sondu alebo gastrostómiu; oneskorený postnatálny vývoj; charakteristické črty tváre; nadmerné skladanie kože; hlboké záhyby kože na dlaniach a chodidlách; hyperpigmentácia kože; prítomnosť papilómov vestibulu nosa a okolo úst; oneskorený intelektuálny vývoj.
Kľúčové slová: deti, Costellov syndróm, dysfágia, nadmerne mäkká pokožka, oneskorený fyzický vývoj.
Autori uvádzajú údaje dostupné z literatúry a ich klinické pozorovanie dieťaťa s Costellovým syndrómom. Pre extrémne zriedkavý genetický syndróm je charakteristická pretrvávajúca dysfágia, ktorá si vyžaduje zavedenie sondy alebo gastrostómiu, spomalenie postnatálneho rastu, charakteristické črty tváre; hyperrugozita kože; hlboké záhyby kože na dlaniach a chodidlách, hyperpigmentácia kože, papilómy nosovej predsiene a okolo úst a mentálna retardácia.
Kľúčové slová: deti, Costellov syndróm, dysfágia, hyperrugozita kože, fyzická retardácia.
Costellov syndróm je extrémne zriedkavé genetické ochorenie, ktoré prvýkrát popísal Dr. D. Costello z Nového Zélandu v roku 1977. V súčasnosti žije na celom svete asi 300 pacientov s týmto syndrómom. Jeho výskyt je 1 z 24 miliónov ľudí, t.j. Na svete sa ročne narodí približne 10 detí s Costellovým syndrómom. Nástup choroby je spojený s mutáciou génu HRAS (chromozomálna lokalizácia 11p13.3), ktorá kóduje syntézu hyperaktívneho proteínu HRAS, ktorý ovplyvňuje rast a delenie buniek. Pri Costellovom syndróme dochádza v tkanivách k nepretržitému aktívnemu deleniu buniek, ktoré prispieva k tvorbe benígnych a malígnych nádorov.
Toto ochorenie sa dedí autozomálne recesívnym spôsobom. K dnešnému dňu veľká časť
Ros Vestn Perinatol Pediat 2010; 5: 27–30
Korešpondenčná adresa: Vasina Tamara Nikolaevna - kandidátka lekárskych vied, docentka, vedúca. oddelenie detské choroby s priebehu detskej chirurgie v Oryol Medical Institute
Zubtsova Tatiana Ivanovna - kandidátka lekárskych vied, doc. oddelenia 302028 Oryol, st. Oktyabrskaya, 4
Stavtseva Svetlana Nikolaevna - genetička v Perinatálnom centre Oryol
Kirsanova Tatyana Aleksandrovna - lekárka ultrazvuková diagnostika Perinatálne centrum Oryol 302019 Oryol, st. Generál Žadov, 4
dovy majú spontánne génové mutácie a možnosť detekcie syndrómu u súrodencov je nízka. Napriek tomu existujú prípady recidívy choroby u bratov a sestier probanda. Prenatálna diagnostika je možná, ak majú členovia rodiny mutáciu v géne HRAS. Väčšina pacientov s Costellovým syndrómom je neplodná.
Diagnóza syndrómu je založená na charakteristických klinických príznakoch a výsledkoch molekulárno-genetických testov. Dôkladné štúdium prenatálnej histórie môže poskytnúť nasledujúce informácie. Pri ultrazvukovom vyšetrení má plod brachycefáliu, skrátenie ramena a stehenná kosť, v 90% prípadov sa vyskytuje polyhydramnión. Popísané rôzne formy predsieňová tachykardia plodu. Väčšina znakov fenotypu plodu je však nešpecifická a otázka prenatálnej diagnostiky zvyčajne nevzniká.
Formálne diagnostické kritériá pre Costellov syndróm ešte neboli vyvinuté, sú však známe hlavné, jedinečné príznaky choroby, vďaka ktorým sú pacienti rozpoznateľní v každom veku. V novorodeneckom období sa pozornosť venuje relatívnej makrocefálii, charakteristickej tvári s veľkými ústami, hrubými perami, nadmerným skladaním kože, širokým nosom, veľkým čelom a epikantom. Najpôsobivejšie klinické
príznakom je nepochybne dysfágia (95% detí), ktorá od narodenia vytvára obrovské problémy s kŕmením, a preto vedie k výraznému zníženiu tempa fyzického vývoja. Pri pokuse o dojčenie dieťaťa alebo cez bradavku má prudkú úzkosť („stresový stav“), odmietnutie klaksónu. Zároveň je zachovaná chuť do jedla a sací reflex. Problémy s kŕmením sú také závažné, že vo väčšine prípadov si vyžadujú nazogastrickú sondu, gastrostómiu alebo Nielsenovu fundoplikáciu. Zároveň môžu deti zostať nosičmi gastrostómie až 4 - 6 rokov.
Medzi bežné znaky treba poznamenať hlboké prehnutie kože na dlaniach a chodidlách, palmárno-plantárna hyperkeratóza, hyperpigmentácia kože v prirodzených záhyboch, pozdĺž stredovej čiary brucha, hyperpigmentácia bradavkovej areoly. Často sú papilómy okolo úst, vo vestibule nosa a perianálne. V 50% prípadov sa zistia rôzne hernie. Upozorňuje sa na výraznú hypotenziu, prudké oneskorenie vo fyzickom, motorickom a neuropsychickom vývoji, úzkosť, podráždenosť.
Už v prvom roku života sa môžu objaviť príznaky stenózy chlopne pľúcnej tepny (dá sa zistiť od narodenia), hypertrofickej kardiomyopatie a supraventrikulárnej tachykardie. Môže sa vytvoriť hydrocefalus; boli opísané epileptické záchvaty.
Deti vo veku od 4 do 12 rokov, ktoré si zachovávajú charakteristické fenotypové znaky, majú nízky rast a ortopedické problémy (kyfoskolióza, torticollis, hypermobilita v kĺboch). V tomto veku začnú samostatne jesť husté aj tekuté jedlo, zbavujú sa tak gastrostómie. Vyznačujú sa svojou spoločenskosťou, priateľskosťou a majú určitý zmysel pre humor. Zjavný je však intelektuálny nedostatok.
Dospievajúci s Costellovým syndrómom majú klasické črty tváre, kučeravé vlasy, nazálnu fibromatózu, hrudné papilómy, hyperkeratózu, hyperpigmentáciu, nízky vzrast, ortopedické poruchy („šľacha s úzkymi pätami“ a deformácie chodidiel) a mentálnu retardáciu. Vyskytuje sa oneskorenie alebo porucha puberty. V dôsledku rastúcej kyfoskoliózy, rednutia vlasov a starnutia pokožky vyzerajú pacienti staršie ako je ich vek. Riziko malígnych novotvarov (rhabdosarkóm, neuroblastóm) je vysoké.
Diferenciálna diagnostika Costellovho syndrómu v dojčenskom veku sa vykonáva pomocou kardiofasciokutánneho syndrómu (CFC), syndrómu Noonana, Beckwitha-Wiedemanna, Simpsona-Galabiho a Williamsa. Podľa odborníkov však fenotyp
costellov syndróm nemožno zamieňať s inou známou génovou poruchou. Detekcia mutácie génu HRAS molekulárnymi genetickými metódami potvrdzuje diagnózu.
Liečba prejavov ochorenia od novorodeneckého obdobia je spojená s poskytovaním adekvátnej výživy, ktorá si najčastejšie vyžaduje použitie nazogastrická sonda a inštalácia gastrostómie. Z dôvodu rozvoja gastroezofageálnej refluxnej choroby je často nutná operácia Nielsenova zábava - doplnenie. Aj keď sa dosiahne príjem kalórií požadovaný vekom, spomalenie rastu pretrváva. Deti so srdcovými problémami sú sledované a liečené kardiológom podľa dostupných štandardov. Deformácie kostry si vyžadujú lekárske fixácie, fyzikálnu terapiu a niekedy chirurgický zákrok. Kognitívne funkcie sa výrazne zlepšia pod vplyvom včasne začatých individuálnych vzdelávacích programov správania. V procese monitorovania pacientov vo vyššom veku môžu byť zistené prípady hypoglykémie. Takéto deti potrebujú dohľad endokrinológa. Na internete existuje webová stránka na kontaktovanie rodín, kde sú vychovávané deti s Costellovým syndrómom.
Uvádzame vlastný prípad klinickej diagnostiky a pozorovania dieťaťa s Costellovým syndrómom.
Dievča R. bolo prijaté na somatické oddelenie regionálnej detskej nemocnice v Orel vo veku 1 mesiaca z dôvodu chudnutia, zadržiavania stolice, úzkosti dieťaťa a ťažkostí s kŕmením.
Anamnéza života a choroby: matka má 30 rokov, považuje seba a otca dieťaťa za zdravé. Dedičnosť sa zhoršuje diabetes mellitus u babičky z matkinej strany. Tehotenstvo 1., žiaduce. Pokiaľ ide o 13 - 14 týždňov tehotenstva, ultrazvukové vyšetrenie odhalilo anechoickú hmotu 7,3 x 5,5 mm na krku, ktorá nesúvisí s chrbticou a kostnou lebkou (krčná cysta?, Nezrelý teratóm?). Ďalej bolo určené rozšírenie hrúbky priestoru goliera až na 2,7 mm. Pri vyšetrení v 14. - 15. týždni tehotenstva bola vpravo identifikovaná aj anechoická hmota krku s rozmermi 5,2 x 5,0 mm. Po opakovaných štúdiách s účasťou genetika v meste Oryol bola žena odoslaná do Výskumného ústavu pôrodníctva a gynekológie (Moskva) s diagnózou podozrenia na cystickú hygromu krku plodu. Vyšetrenie sérových markerov prenatálnej fetálnej patológie uskutočnené do 16 týždňov tehotenstva ukázalo pokles hladiny a-fetoproteínu na 0,43 Mot. Bolo rozhodnuté o predĺžení tehotenstva.
V 19. týždni tehotenstva sa predpokladalo, že existujú filiálne sínusy plodu, ktoré sa neskôr vizualizovali. Ultrazvukové vyšetrenie v 23. - 24. týždni tehotenstva odhalilo polyhydramnión, čo naznačuje zníženie prehĺtania
Vasina T.N. a kol. Klinická diagnóza zriedkavého dedičného ochorenia - Costellov syndróm
činnosť plodu. Od 24. týždňa tehotenstvo pokračovalo so známkami uteroplacentárnej nedostatočnosti, hyperpláziou placenty; v čase 29., 31., 37. týždňa tehotenstva bola žena liečená v nemocnici na pyelonefritídu.
Prvé pôrody v 38-39 týždni tehotenstva. Trvanie prvej menštruácie je 9 hodín; II obdobie - 20 minút, bezvodý interval - 4 hodiny 50 minút. Zaznamenal sa polyhydramnión, plodová voda bola zelená, zadná strana bola hnedozelená. V súvislosti s aspiráciou plodovou vodou, horná dýchacích ciest, umelá ventilácia pľúc po dobu 13 min. Apgar skóre 7/8 bodov, pôrodná hmotnosť 2980 g, dĺžka 49 cm, obvod hlavy 34 cm V pôrodnici boli zaznamenané príznaky depresívneho syndrómu CNS a viacnásobných stigiem dysembryogenézy, bola vykonaná infúzia a patogenetická terapia.
V 5. deň svojho života bolo dievča preložené na oddelenie patológie novorodencov Detskej regionálnej nemocnice v Orel, kde bolo 2 týždne liečené s diagnózou perinatálne hypoxické poškodenie mozgu, cerebrálna ischémia II. Stupňa, depresívny syndróm; intrauterinná retardácia rastu hypotrofického typu; viacnásobné stigmy dysembryogenézy. Dieťa malo výraznú hypersoftnú pokožku tváre a krku; hlboký skladací a priečny pruh na chodidlách; hypertelorizmus palpebrálnych trhlín; veľké ústa; široký mostík nosa; dysmorfizmus ušných boltcov; hemangiómy pokožky hlavy a zadnej časti krku; krátky krk (pozri obrázok). Konzultoval ju s genetikom, bolo podozrenie na Shere-Shevsky-Turnerov syndróm. Vykonaným karyotypom sa zistil normálny karyotyp 46, XX. Dodatočné ultrazvukové vyšetrenie odhalilo príznaky hypoxicko-ischemického poškodenia mozgu (bočné komory s priemerom 2,6 mm); pyeloektázia vľavo 6,3 mm.
Podstúpené ošetrenie: kŕmenie zmesou „Nan“, vitamínu B6, intramuskulárneho ATP, cavinton, espu-mizan, orálny bifidumbacterín, masáž. Dieťa sa prisalo pomaly, ale nevracalo sa, stolica bola pravidelná. S telesnou hmotnosťou 2920 g bola prepustená pod ambulantným dohľadom miestneho pediatra a neuropatológa.
Doma matka čelila pri kŕmení dieťaťa vážnym problémom: po niekoľkých sacích pohyboch sa dievča začalo prudko trápiť, odvrátilo sa od bradavky a bránilo obnoveniu kŕmenia. Po chvíli na chvíľu zaspala, potom sa zobudila, opäť začala nenásytne sať a rovnako náhle prestala kŕmiť. Nezávislé kreslo postupne zaniklo. Vo veku 1 mesiaca po stretnutí s pediatrom v poliklinike bola telesná hmotnosť 2700 g. S diagnózou podvýživy III. Stupňa bolo dieťa poslané do detského areálu.
Obrázok. Novorodenec s Costellovým syndrómom.
nová nemocnica.
Stav pri prijatí bol ťažký z dôvodu silnej hypotrofie. Pri vyšetrení je negatívny, krik je hlasný, prenikavý. Upokojí sa s cumlíkom, energicky saje. Z fľaše začne zmes a voda aktívne sať, nie však viac ako 10 ml, potom dôjde k ostrej úzkosti, plaču, úniku z bradavky. Regurgitácia a zvracanie sa nepozorovali. Keď bolo dieťa pozorované, dojem prítomnosti dysfágie sa ustálil.
Dievčatko malo zvláštne fenotypové znaky: bolo neprimerané vzhľadom na pomerne veľkú veľkosť hlavy (obvod 36,5 cm), telo a končatiny boli tenké. Koža je suchá, svetlo bledá, s nadmernými záhybmi na tvári a krku; hlboké záhyby kože na dlaniach a chodidlách. Areoly bradaviek, stredná čiara brucha, pupočníkový krúžok, prirodzené záhyby sú hyperpigmentované. Existuje pupočná kýla a kýla strednej čiary brucha. Očné štrbiny s epikantom a hypertelorizmom, občas zbiehavé zášklby. Široký mostík nosa, vysoké „gotické“ podnebie, kapilárne
hemangióm predsiene nosa, nasolabiálny trojuholník. Ušnice sú fantasticky stočené, nastavené nízko. Veľká fontanela 2x2 cm, trochu sa potápa. Dýchanie dieťa, žiadna dýchavičnosť. Zvuky srdca sú hlasné, žiadne šelesty, pulz 140 za minútu. Brucho je mäkké, pečeň a slezina sú na okraji pobrežnej klenby citeľné. Stolica po klystíre je kašovitá, sporá. Pohlavné orgány sa tvoria podľa ženského typu. Neurologický stav: semi-flexné držanie tela, stredná spontánna motorická aktivita, tremor rúk, svalová dystónia v končatinách.
Všeobecne a biochemické krvné testy patologické zmeny nebolo zaznamenané. Podľa údajov z ultrazvuku sa pečeň, slezina, žalúdok, nadobličky nemenia; pyelektáza vľavo 7,8 mm. Neurosonografia: laterálne komory po 3,5 mm, interhemisférická medzera 2 mm, neboli zistené patologické zmeny v mozgu. Oculistická patológia nebola stanovená. Lekár ORL zaznamenal anatomicky krátky krk, úzky nosohltan, vysoké postavenie epiglottis. Echokardiografia neodhalila žiadne anatomické chyby. Po konzultácii s chirurgom s cieľom objasniť príčiny dysfágie a vylúčiť vrodené malformácie gastrointestinálneho traktu bola vykonaná ezofagogastroduodenoskopia, ktorá neodhalila žiadnu organickú patológiu. Kontrastná fluoroskopia pažeráka, žalúdka, čriev: obraz kardiospazmu, pylorospazmu; evakuácia zo žalúdka je spomalená. Liečba sa uskutočňovala: kŕmenie zmesou Alfare po 3 hodinách pomocou nazogastrickej sondy, espumisanu, motilium, el.
auto, kreon, bifidumbacterin.
Dieťa bolo opätovne konzultované s genetikom, na základe klinických údajov sa predpokladalo, že je prítomný Costellov syndróm. Odporúčala sa konzultácia a možné vyšetrenie v Centre lekárskeho genetického výskumu Ruskej akadémie lekárskych vied. Dievčatko bolo prepustené so zlepšením stavu pod dohľadom miestneho pediatra, neuropatológa, genetika s odporúčaním na kŕmenie hadičkou so zmesami na báze proteínových hydrolyzátov a na liečbu energotropnou terapiou. Ďalšie dynamické pozorovanie dieťaťa umožňuje stanoviť predpokladanú diagnózu, pretože existujú hlavné diagnostické príznaky choroby:
Dysfágia;
Oneskorenie tempa fyzického vývoja;
Hlboká retardácia psychomotorického vývoja;
Hyperskladanie čela;
Hlboké kožné záhyby na dlaniach a chodidlách;
Hyperpigmentácia pupočníka;
Hemangióm predsiene nosa;
Charakteristické črty tváre (hypertelorizmus očných štrbín, hrubé, vtlačené pery, veľké ústa, jazyk, široký nosový mostík);
Vznikajúci hydrocefalický syndróm.
V domácej literatúre sa nám nepodarilo nájsť správy o pozorovaniach pacientov s Costellovým syndrómom. Predložený prípad klinickej diagnostiky zriedkavého genetického ochorenia je jedinečný a je vedecky i prakticky zaujímavý pre lekárov rôznych špecializácií.
LITERATÚRA
1. Gripp K.W., Lin A.E., Stabley D.L. a kol. Analýza mutácií HRAS pri Costellovom syndróme: genotypová a fenotypová korelácia // Am. J. Med. Genet. 2006. Zv. 140A. S. 1-7.
2. Kozlova S.I., Demikova N.S., Semanova E, Blinnikova O.E. Dedičné syndrómy a lekárske genetické poradenstvo. M.: Practice, 1996. S. 122-123.
3. Gripp K. W., Lin A. E., Nicholson L. a kol. Ďalšie vymedzenie fenotypu, ktorý je výsledkom zárodočnej línie BRAForMEKI
mutácia pomáha odlíšiť kardio-facio-cutaneos syndróm od Costellovho syndrómu // Am. Y. Med. Genet. 2007. Zv. 143A. P. 1472-1480.
4. Kerr B, Delrue M.-A., Sigaudy S. a kol. Korelácia genotyp-fenotyp pri Costellovom syndróme: analýza mutácie HRAS v 43 prípadoch // J. Med. Genet. 2006. Zv. 43. S. 401-405.
- Yoon S, Seger R. Extracelulárna signálne regulovaná kináza: Viacero substrátov reguluje rôzne bunkové funkcie. Rastové faktory. 2006; 24 (1): 21-44. doi: 10.1080 / 02699050500284218
- Rauen KA. The RASopathies. Ročný prehľad genomiky a genetiky človeka. 2013; 14 (1): 355-369. doi: 10,1146 / annurev-genom-091212-153523
- Bos JL. RAS onkogény pri rakovine človeka: prehľad. Výskum rakoviny. 1989 1. september 1989; 49 (17): 4682-4689.
- Carragher L, Pritchard C, Aldridge V, Giblett S, Jin H, Foster C a kol. Myšie modely pre rakovinu indukovanú BRAF. Transakcie s biochemickou spoločnosťou. 2007; 35 (5): 1329. doi: 10,1042 / bst0351329
- Wallace M, Marchuk D, Andersen L, Letcher R, Odeh H, Saulino A a kol. Gén neurofibromatózy typu 1: identifikácia veľkého prepisu narušeného u troch pacientov s NF1. Veda. 1990; 249 (4965): 181-186. doi: 10,1126 / science.2134734
- Čižmárová M, Kostalová L, Pribilincová Z, Lasabová Z, Hlavatá A, Kovács L a kol. Rasopatie - dysmorfické syndrómy s nízkym vzrastom a rizikom malignity. Endokrinné predpisy. 2013; 47 (04): 217-222. doi: 10,4149 / endo_2013_04_217
- Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Rakovina v syndrómoch Noonan, Costello, kardiofaciokutánnom a LEOPARD. American Journal of Medical Genetics Časť C: Semináre z lekárskej genetiky. 2011; 157 (2): 83-89. doi: 10,1002 / ajmg.c.30300
- Tartaglia M, Gelb BD, Zenker M. Noonanov syndróm a klinicky súvisiace poruchy. Najlepšie postupy a výskum Klinická endokrinológia a metabolizmus. 2011; 25 (1): 161-179. doi: 10.1016 / j.beem.2010.09.002
- Malaquias AC, Brasil AS, Pereira AC, Arnhold IJP, Mendonca BB, Bertola DR a kol. Rastové štandardy pacientov s Noonanom a syndrómmi podobnými Noonanovi s mutáciami v dráhe RAS / MAPK. American Journal of Medical Genetics Part A. 2012; 158A (11): 2700-2706. doi: 10,1002 / ajmg.a,35519
- Noordam K, van der Bürgt I, Brunner HG, Otten BJ. Vzťah medzi klinickou závažnosťou Noonanovho syndrómu a rastom, sekréciou rastového hormónu (GH) a reakciou na liečbu GH. Journal of Pediatric Endocrinology and Metabolism. 200; 15 (2). Doi: 10,1515 / jpem.2002.15.2.175
- Binder G, Neuer K, Ranke MB, Wittekindt NE. Mutácie PTPN11 sú spojené s miernou rezistenciou rastového hormónu u jedincov s Noonanovým syndrómom. The Journal of Clinical Endocrinology & Metabolism. 2005; 90 (9): 5377-5381. doi: 10.1210 / jc.2005-0995
- van der Burgt I. Orphanet Journal of Rare Diseases. 2007; 2 (1): 4. doi: 10,1186 / 1750-1172-2-4
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H a kol. Mutácie v PTPN11 kódujúce proteínovú tyrozínfosfatázu SHP-2 spôsobujú Noonanov syndróm. Genetika prírody. 2001; 29 (4): 465-468. doi: 10,1038 / ng772
- Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A a kol. Mutácie SOS1 so zosilnením funkcie spôsobujú výraznú formu Noonanovho syndrómu. Genetika prírody. 2006; 39 (1): 75-79. doi: 10,1038 / ng1939
- Zenker M, Horn D, Wieczorek D, Allanson J, Pauli S, van der Burgt I a kol. SOS1 je druhý najbežnejší gén Noonan, ale nehrá žiadnu významnú úlohu pri kardio-facio-kožnom syndróme. Journal of Medical Genetics. 2007; 44 (10): 651-656. doi: 10,1136 / jmg.2007.051276
- Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S a kol. Mutácie RAF1 so zosilnením funkcie spôsobujú syndrómy Noonan a LEOPARD s hypertrofickou kardiomyopatiou. Genetika prírody. 2007; 39 (8): 1007-1012. doi: 10,1038 / ng2073
- Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R a kol. Mutácie zárodočnej funkcie zárodočnej línie v RAF1 spôsobujú Noonanov syndróm. Genetika prírody. 2007; 39 (8): 1013-1017. doi: 10,1038 / ng2078
- Gremer L, Merbitz-Zahradnik T, Dvorský R, Cirstea IC, Kratz CP, Zenker M a kol. Zárodočné mutácie KRAS spôsobujú abnormálne biochemické a fyzikálne vlastnosti vedúce k vývojovým poruchám. Ľudská mutácia. 2011; 32 (1): 33-43. doi: 10,1002 / humu.21377
- Cirstea IC, Kutsche K, Dvorský R, Gremer L, Carta C, Horn D a kol. Obmedzené spektrum mutácií NRAS spôsobuje Noonanov syndróm. Genetika prírody. 2009; 42 (1): 27-29. doi: 10,1038 / ng.497
- Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F a kol. GermlineBRAF mutácie v Noonan, LEOPARD a kardiofaciokutánnych syndrómoch: Molekulárna diverzita a súvisiace fenotypové spektrum. Ľudská mutácia. 2009; 30 (4): 695-702. doi: 10,1002 / humu.20955
- Komatsuzaki S, Aoki Y, Niihori T, Okamoto N, Hennekam RCM, Hopman S a kol. Mutačná analýza génu SHOC2 pri Noonanovom syndróme a pri hematologických malignitách. Journal of Human Genetics. 2010; 55 (12): 801-809. doi: 10.1038 / jhg.2010.116
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma "ayan A, Sarkozy A, Fodale V a kol. Mutácia SHOC2 podporuje aberantnú bielkovinu N-myristoyláciu a spôsobuje syndróm podobný Noonanovi s rozpustenými anagénnymi vlasmi. Genetika prírody. 2009; 41 (9): 1022-1026. doi: 10,1038 / ng.425
- Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, Lepri F a kol. Heterozygotné mutácie zárodočnej línie v géne na potlačenie nádoru CBL spôsobujú fenotyp podobný noonanovému syndrómu. American Journal of Human Genetics. 2010; 87 (2): 250-257. doi: 10.1016 / j.ajhg.2010.06.015
- Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ a kol. Zárodočné mutácie CBL spôsobujú vývojové abnormality a predisponujú k juvenilnej myelomonocytovej leukémii. Genetika prírody. 2010; 42 (9): 794-800. doi: 10,1038 / ng.641
- Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K a kol. Mutácie zosilnenia funkcie v RIT1 spôsobujú Noonanov syndróm, syndróm RAS / MAPK Pathway. American Journal of Human Genetics. 2013; 93 (1): 173 - 180. doi: 10.1016 / j.ajhg.2013.05.021
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatóza typu 1 znovu navštívená. Pediatria. 2009; 123 (1): 124-133. doi: 10,1542 / peds.2007-3204
- Sarkozy A, Digilio M, Dallapiccola B. Leopardí syndróm. Orphanet Journal of Rare Diseases. 2008; 3 (1): 13. doi: 10,1186 / 1750-1172-3-13
- Revencu N, Boon LM, Mendola A, Cordisco MR, Dubois J, Clapuyt P a kol. RASA1 Mutácie a súvisiace fenotypy u 68 rodín s kapilárnou malformáciou - arteriovenóznou malformáciou. Ľudská mutácia. 2013; 34 (12): 1632-1641. doi: 10,1002 / humu.22431
- Boon LM, Mulliken JB, Vikkula M. RASA1: variabilný fenotyp s kapilárnymi a arteriovenóznymi malformáciami. Súčasné stanovisko v genetike a vývoji. 2005; 15 (3): 265-269. doi: 10.1016 / j.gde.2005.03.004
- Rauen KA. HRAS a Costellov syndróm. Klinická genetika. 2007; 71 (2): 101-108. doi: 10.1111 / j.1399-0004.2007.00743.x
- Siegel DH, Mann JA, Krol AL, Rauen KA. Dermatologický fenotyp pri Costellovom syndróme: dôsledky vývojovej dysregulácie Ras. British Journal of Dermatology. 2012; 166 (3): 601-607. doi: 10.1111 / j.1365-2133.2011.10744.x
- Niihori T, Aoki Y, Narumi Y, Neri G, Cavé H, Verloes A a kol. Mutácie zárodočnej KRAS a BRAF pri kardio-fakio-kožnom syndróme. Genetika prírody. 2006; 38 (3): 294-296. doi: 10,1038 / ng1749
- Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. Fosfatázový holoenzým zložený z Shoc2 / Sur8 a katalytickej podjednotky funkcií PP1 ako M-Ras efektor na moduláciu aktivity Raf. Molekulárna bunka. 2006; 22 (2): 217-230. doi: 10.1016 / j.molcel.2006.03.027
- Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T a kol. Kardio-faciokutánne a Noonanove syndrómy v dôsledku mutácií v signálnej ceste RAS / MAPK: vzťahy fenotypu a genotypu a prekrývajú sa s Costellovým syndrómom. Journal of Medical Genetics. 2007; 44 (12): 763-771. doi: 10,1136 / jmg.2007.050450
- Yoon G, Rosenberg J, Blaser S, Rauen KA. Neurologické komplikácie kardio-facio-kožného syndrómu. Vývojová medicína a detská neurológia. 2007; 49 (12): 894-899. doi: 10.1111 / j.1469-8749.2007.00894.x
- Brems H, Legius E. Legiov syndróm, aktualizácia, molekulárna patológia mutácií v SPRED1. The Keio Journal of Medicine. 2013; 62 (4): 107-112. doi: 10,2302 / kjm.2013-0002-RE
- Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA a kol. Mutácie zárodočnej funkcie zárodočnej línie v SOS1 spôsobujú Noonanov syndróm. Genetika prírody. 2006; 39 (1): 70-74. doi: 10,1038 / ng1926
- Narumi Y, Aoki Y, Niihori T, Sakurai M, Cavé H, Verloes A a kol. Klinické prejavy u pacientov s mutáciami SOS1 sa pohybujú od Noonanovho syndrómu po CFC syndróm. Journal of Human Genetics. 2008; 53 (9): 834-841. doi: 10,1007 / s10038-008-0320-0
- Fabretto A, Kutsche K, Harmsen M-B, Demarini S, Gasparini P, Fertz MC a kol. Dva prípady Noonanovho syndrómu so závažným respiračným a gastroenterálnym postihnutím a mutáciou SOS1 F623I. Európsky vestník lekárskej genetiky. 2010; 53 (5): 322-324. doi: 10.1016 / j.ejmg.2010.07.011
- Wennerberg K. Nadrodina RAS v skratke. Journal of Cell Science. 2005; 118 (5): 843-846. doi: 10,1242 / jcs.01660
- Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F a kol. Kraniosynostóza u pacientov s Noonanovým syndrómom spôsobená zárodočnými mutáciami KRAS. American Journal of Medical Genetics Part A. 2009; 149A (5): 1036-1040. doi: 10,1002 / ajmg.a.32786
- Stark Z, Gillessen-Kaesbach G, Ryan MM, Cirstea IC, Gremer L, Ahmadian MR a kol. Dve nové zárodočné mutácie KRAS: rozšírenie molekulárneho a klinického fenotypu. Klinická genetika. 2012; 81 (6): 590-594. doi: 10.1111 / j.1399-0004.2011.01754.x
- Oliveira JB, Bidere N, Niemela JE, Zheng L, Sakai K, Nix CP a kol. Mutácia NRAS spôsobuje ľudský autoimunitný lymfoproliferatívny syndróm. Zborník prác Národnej akadémie vied. 2007; 104 (21): 8953-8958. doi: 10,1073 / pnas.0702975104
- Kobayashi T, Aoki Y, Niihori T, Cavé H, Verloes A, Okamoto N a kol. Molekulárna a klinická analýza RAF1in Noonanovho syndrómu a príbuzných porúch: defosforylácia serínu 259 ako základného mechanizmu aktivácie mutantov. Ľudská mutácia. 2010; 31 (3): 284-294. doi: 10,1002 / humu.21187
- Tidyman WE, Rauen KA. RASopathies: vývojové syndrómy dysregulácie dráhy Ras / MAPK. Súčasné stanovisko v genetike a vývoji. 2009; 19 (3): 230-236. doi: 10.1016 / j.gde.2009.04.001
- Swaminathan G, Tsygankov AY. Proteíny rodiny Cbl: Vodcovia kruhu v regulácii bunkovej signalizácie. Časopis bunkovej fyziológie. 2006; 209 (1): 21-43. doi: 10,1002 / jcp.20694
- Flynn DC. Adaptérové \u200b\u200bproteíny. Onkogén. 2001; 20 (44): 6270-6272. doi: 10,1038 / sj.onc.1204769
- Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernandez H a kol. Spoločný molekulárny mechanizmus je základom ľudských rasopatií Legiovho syndrómu a neurofibromatózy-1. Gény a vývoj. 2012; 26 (13): 1421-1426. doi: 10,1101 / gad.190876.112
Costellov syndróm je veľmi zriedkavé ochoreniektorý ovplyvňuje niekoľko telesných systémov, spôsobuje nízky vzrast, charakteristické črty tváre, výrastky okolo nosa a úst a srdcové problémy. Príčina Costellovho syndrómu nie je známa, aj keď je podozrenie na genetickú mutáciu. V roku 2005 vedci z Detskej nemocnice DuPont v Delaware (USA) zistili, že génové mutácie v sekvencii HRAS boli prítomné u 82,5% zo 40 ľudí s Costellovým syndrómom, ktorí študovali.
V globálnej lekárskej literatúre bolo publikovaných iba asi 150 správ o Costellovom syndróme, takže nie je jasné, ako často sa tento syndróm skutočne vyskytuje alebo kto je pravdepodobnejšie postihnutý.
príznaky
Typické príznaky Costellovho syndrómu:
- Ťažkosti s priberaním a pribúdaním po narodení, ktoré majú za následok nízky vzrast
- Nadmerne voľná pokožka na krku, dlaniach, prstoch a chodidlách (cutis laxa)
- Nezhubné novotvary (papilómy) okolo úst a nosných dierok
- Výrazný vzhľad tváre, ako napríklad veľká hlava, nízko nasadené uši s veľkými, hrubými okvetnými lístkami, hrubými perami a / alebo širokými nozdrami
- Mentálna retardácia
- Zahustená, suchá pokožka rúk a nôh alebo rúk a nôh (hyperkeratóza)
- Abnormálne pružné kĺby prstov.
Niektorí ľudia môžu mať obmedzený pohyb v lakťoch alebo stlačenie šľachy v zadnej časti členka. Ľudia s Costellovým syndrómom môžu mať srdcové chyby alebo srdcové choroby (kardiomyopatia). Existuje vysoký výskyt nádorového rastu, malígneho aj nezhubného, \u200b\u200btiež spojený so syndrómom.
diagnostika
Diagnóza Costellovho syndrómu je založená na vzhľad dieťa narodené s poruchou, ako aj ďalšie príznaky, ktoré sa môžu vyskytnúť. Väčšina detí s Costellovým syndrómom má ťažkosti s kŕmením a tiež priberajú a rastú, takže to môže naznačovať diagnózu. Nabudúce genetické testovanie pre známe génové mutácie spojené s Costellovým syndrómom možno použiť na potvrdenie diagnózy.
liečby
Pre Costellov syndróm neexistuje žiadna špecifická liečba, preto sa lekárska starostlivosť zameriava na existujúce príznaky a poruchy. Všetkým ľuďom s Costellovým syndrómom sa odporúča podstúpiť srdcové vyšetrenie na kontrolu srdcových chýb alebo srdcových chorôb. Fyzikálna a pracovná terapia môže pomôcť človeku dosiahnuť jeho vývojový potenciál. Dôležitosť má dlhodobé sledovanie rastu nádorov, problémov s chrbticou alebo ortopedom a zmien srdca alebo krvného tlaku. Očakávaná dĺžka života osoby s Costellovým syndrómom bude ovplyvnená prítomnosťou srdcových problémov alebo rakoviny, takže ak sú zdraví, ľudia so syndrómom môžu mať normálnu dĺžku života.
Costellov syndróm je zriedkavá genetická porucha spojená s oneskoreným fyzickým a duševným vývojom.
Ovplyvňuje rôzne časti tela a vyznačuje sa uvoľnenými kožnými záhybmi, zlým svalovým tonusom a inými problémami.
Medzi ďalšie komplikácie patrí vývoj zhubných a nezhubných nádorov, srdcové chyby a abnormálny rast srdcového svalu.
Medzi bežné problémy so srdcom patrí hypertrofická kardiomyopatia, čo je zväčšenie srdca, ktoré oslabuje srdcový sval, abnormálne srdcové rytmy alebo arytmie a ďalšie štrukturálne chyby.
Predpokladá sa, že Costellov syndróm, tiež známy ako Faciocutaneoskeletálny syndróm (FCS), postihuje 200-300 ľudí na celom svete, ale viac prípadov môže zostať nediagnostikovaných.
Rýchle fakty o Costellovom syndróme
- Costellov syndróm je extrémne zriedkavý a postihuje 200 až 300 ľudí na celom svete.
- To môže viesť k oneskoreniu vývoja, mentálnemu poškodeniu, veľkej hlave a perám s nízkymi ušami a uvoľnenej pokožke.
- Costellov syndróm tiež spôsobuje problémy so srdcom. Jedná sa o genetický stav, ktorý ovplyvňuje množstvo telesných systémov.
- Neexistuje žiadna pokračujúca liečba a žiadne špeciálne postupy pre tento stav. Liečba je zameraná na zmiernenie rôznych aspektov syndrómu, ako je hypertrofická kardiomyopatia a špeciálne vzdelávanie na podporu porúch učenia.
príznaky
Väčšina príznakov Costellovho syndrómu nie je prítomná pri narodení, ale objavia sa, keď dieťa začne rásť.
Pôrodná hmotnosť je zvyčajne normálna alebo mierne nadpriemerná, ale dojča bude rásť pomalšie ako väčšina detí.
Medzi príznaky patria:
- malá výška a pomalý rast
- Mentálne postihnutie
- vývojové oneskorenie
- ťažkosti s dojčením
- veľká hlava
- voľná pokožka, najmä na rukách a nohách
- hlboké záhyby v dlaniach a chodidlách
- nízke uši, silné ušné laloky alebo oboje
- flexibilné spojenia
- veľké ústa
- povrch tváre sa javí drsný
- strabizmus
- srdcové problémy vrátane abnormálneho srdcového rytmu
- problémy so zubami
- hustá achilová šľacha
- hrubé mozole a nechty na nohách
komplikácie
Costellov syndróm je komplexný multisystémový stav, ktorý môže viesť k rôznym komplikáciám.
Bábätká sa nemôžu orálne kŕmiť, kým nedosiahnu vek 2 až 4 rokov alebo približne v rovnakom čase, keď začnú rozprávať.
Kardiovaskulárne problémy sa často objavujú už od raného detstva, aj keď príznaky sa nemusia prejaviť neskôr. Tie obsahujú vrodené chyby srdce a hypertrofia srdca. U osoby sa môžu vyskytnúť tachykardia alebo rýchle srdcové rytmy, arytmie alebo nepravidelné srdcové rytmy.
Makrocefália alebo nadmerný rast mozgu bol zaznamenaný u 50% pacientov. To podľa jednej štúdie môže viesť k Chiariho malformácii, štrukturálnej poruche mozgu zistenej u 32 percent ľudí. Zdá sa, že záchvaty postihujú 20 až 50 percent ľudí s týmto ochorením.
Možno pozorovať spomalenie rastu kostí, nízku hustotu kostí a vyššie riziko zlomenín a osteoporózy. Pomôcť môžu vitamíny D a vápnik.
Môžu sa vyvinúť nádory, najčastejšie papilómy, malé výrastky, ktoré sa podobajú bradaviciam, najmä okolo nosa, úst a konečníka.
Ľudia s Costellovým syndrómom sú náchylní na vývoj rakovinových aj nerakovinových nádorov. Rakoviny zahŕňajú rabdomyosarkóm, neuroblastóm a prechodný bunkový karcinóm.
Dva ďalšie genetické stavy s podobnými príznakmi sú Noonanov syndróm a kardiovaskulárny syndróm (CFC). Prekrývajúce sa príznaky sťažujú diagnostiku Costellovho syndrómu v detstve.
dôvody
Costellov syndróm je genetická porucha spôsobená mutáciami v géne HRAS. Toto je gén, ktorý dáva telu pokyn, aby vytvoril proteín známy ako H-Ras. H-Ras podporuje rast a delenie buniek.
Mutácie v géne HRAS, ktoré sa vyskytujú pri Costellovom syndróme, spôsobujú, že bunky neustále rastú a delia sa, a to nielen na základe pokynov.
To môže viesť k rakovinovému aj nerakovinovému množeniu nádoru a je pravdepodobné, že bude podkladom pre ďalšie príznaky.
Mutácia génu HRAS môže tiež ovplyvniť produkciu elastických vlákien v tkanive. Tieto vlákna sú životne dôležité pre štruktúry ako sú pľúca, pokožka a veľké krvné cievy vrátane aorty.
Vláknina je dôležitá pre udržanie silných vlasov a pokožky, prevenciu raného sexu a pre udržanie celistvosti a sily krvných ciev a pľúcneho tkaniva.
Aby sa vyvinul Costello syndróm, musí sa dediť iba jedna kópia mutovaného génu HRAS. Tento typ dedičnosti génov je známy ako autozomálna dominancia.
Väčšina prípadov Costellovho syndrómu je spojená s novými mutáciami, pri ktorých nie je v rodinnej anamnéze uvedený stav.
diagnostika
Costellov syndróm je veľmi zriedkavý, takže je nepravdepodobné, že ho lekár okamžite podozrie.
Lekár najskôr začne s hodnotením výšky, veľkosti hlavy a pôrodnej hmotnosti dieťaťa.
Ďalšia etapa zahŕňa molekulárne genetické testovanie. Vykoná sa sekvenčná analýza génu HRAS, aby sa zistilo, či existuje mutácia spojená s Costellovým syndrómom.
liečby
Neexistuje žiadny liek na Costellov syndróm ani žiadna konkrétna liečba, ale aspekty syndrómu, ako sú srdcové choroby, hypertrofická kardiomyopatia, sa dajú špecificky liečiť.
Spôsoby, ako pomôcť človeku zvládnuť tento stav:
- pomáha dieťaťu prekonať ťažkosti s kŕmením počas dojčenia
- liečbe srdcových problémov
- poskytovanie špeciálneho vzdelávania
Vedci hľadajú efektívna metóda liečba stavu na genetickej úrovni.
Medzi ďalšie aktivity patrí:
- profesionálna a fyzioterapia
- chirurgický zákrok na predĺženie achilovky
- odstránenie papilómov pomocou kryoterapie
predpoveď
Očakávaná dĺžka života niekoho s Costellovým syndrómom nebola formálne študovaná. Výskum ukazuje, že ak dôjde k úmrtiu, môže to byť dôsledok komplikácií, ako je napríklad problém so srdcom.