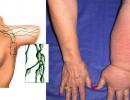
Kostello sindromo aprašymas. Naujagimio retos ligos atvejis. Tatarstano Respublikos sveikatos apsaugos ministerijos vaikų Respublikinė klinikinė ligoninė
Kostello sindromas taip pat vadinama faziocutaneoskeletinis sindromas arba fCS sindromas , yra retas genetinis sutrikimas, paveikiantis daugelį kūno dalių. Jam būdingas vystymosi ir protinis atsilikimas, išskirtiniai veido bruožai, neįprastai lankstūs sąnariai ir laisvi papildomos odos raukšlės, ypač ant rankų ir kojų. Širdies sutrikimai yra dažni, įskaitant labai greitą širdies plakimą (tachikardija), širdies struktūrinius defektus ir širdies raumens padidėjimą (hipertrofinė kardiomiopatija). Vaikų, sergančių „Costello“ sindromu, gimimo metu gali būti daug, tačiau jie auga lėčiau nei kiti vaikai ir turi mitybos sunkumų. Vėliau šios ligos žmonės turi palyginti trumpą ūgį ir daug sumažėjusį augimo hormonų kiekį. Tai RASopatija.
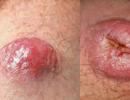
Nuo ankstyvos vaikystės žmonėms, sergantiems Costello sindromu, yra padidėjusi rizika susirgti tam tikrais vėžiniais ir vėžiniais navikais. Maži augliai, vadinami papilomais, yra dažniausiai pasitaikantys gerybiniai navikai, pastebėti esant šiai būklei. Paprastai jie išsivysto aplink nosį ir burną arba šalia išangės. Dažniausias vėžinis navikas, susijęs su Costello sindromu, yra minkštųjų audinių navikas, vadinamas rabdomiosarkoma. Taip pat pranešta apie kitus vėžius, sergančius šia liga sergantiems vaikams ir paaugliams, įskaitant naviką, atsirandantį dėl nervinių ląstelių vystymosi (neuroblastoma), ir šlapimo pūslės vėžio formą (pereinamojo laikotarpio ląstelių karcinoma).
Kostello sindromą atrado daktaras Jackas Costello, Naujosios Zelandijos pediatras 1977 m., Ir jis yra įskaitytas į pirmuosius sindromo pranešimus 1977 m. Australijos žurnale Pediatrics, 13 tomas, Nr. 2.
ženklai ir simptomai
genetika
Kostello sindromą sukelia bent viena iš penkių skirtingų mutacijų HRAS genas 11 chromosomoje. Šiame gene yra nurodymai, kaip pagaminti baltymą H-Ras, padedantį kontroliuoti ląstelių augimą ir dalijimąsi. Costello sindromą sukeliančios mutacijos lemia H-Ras baltymo, kuris yra nuolat aktyvus, gamybą. Hiperaktyvus baltymas, užuot suaktyvinęs ląstelės augimą, reaguodamas į tam tikrus signalus iš ląstelės išorės, nurodo ląstelei nuolat augti ir dalintis. Tai, kad nepavyksta sustabdyti ląstelių dalijimosi, gali sukelti pacientams gerybinių ir piktybinių navikų vystymąsi. Lieka neaišku, kaip mutacijos HRAS sukelia kitus Costello sindromo požymius, tačiau daugybė požymių ir simptomų gali atsirasti dėl ląstelių dauginimosi ir nenormalios ląstelių dalijimosi.
Klinikinė retos paveldimos ligos diagnozė - Costello sindromas
T.N. Vasina, T.I. Zubtsova, S.N. Stavtseva, T.A. Kirsanova
Klinikinė retos paveldimos ligos diagnozė: Costello sindromas
T.N. Vasina, T.I. Zubtsova, S.N. Stavtseva, T.A. Kirsanova
Orilo medicinos institutas
Pateikti literatūros duomenys ir paties vaiko, sergančio Costello sindromu, klinikiniai stebėjimai. Ypač retam genetiniam sindromui būdinga nuolatinė disfagija, reikalaujanti vamzdelio ar gastrostominio vamzdelio; uždelstas vystymasis po gimdymo; būdingi veido bruožai; per didelis odos sulankstymas; gilūs odos raukšlės ant delnų ir padų; odos hiperpigmentacija; nosies vestibiulės ir aplink burną esančių papilomų buvimas; uždelstas intelekto vystymasis.
Raktažodžiai: vaikai, Costello sindromas, disfagija, hipersoft oda, uždelstas fizinis vystymasis.
Autoriai pateikia literatūroje prieinamus duomenis ir klinikinį vaiko, sergančio Costello sindromu, stebėjimą. Itin retam genetiniam sindromui būdinga nuolatinė disfagija, dėl kurios reikia įkišti vamzdelį ar atlikti gastrostomiją, po gimdymo sulėtėjęs augimas, būdingi veido bruožai; odos hipergrozumas; gilūs odos raukšliai delnuose ir paduose, odos hiperpigmentacija, nosies vestibiulio ir aplink burną esančios papilomos ir intelekto atsilikimas.
Raktažodžiai: vaikai, Costello sindromas, disfagija, odos hipergruzitetas, fizinis atsilikimas.
Kostello sindromas yra ypač reta genetinė liga, kurią pirmą kartą aprašė gydytojas D. Costello iš Naujosios Zelandijos 1977 m. Šiuo metu pasaulyje yra apie 300 šiuo sindromu sergančių pacientų. Jos pasireiškia 1 iš 24 milijonų žmonių, t. Kiekvienais metais visame pasaulyje gimsta apie 10 vaikų, sergančių „Costello“ sindromu. Ligos pradžia siejama su HRAS geno mutacija (chromosomų lokalizacija 11p13.3), koduojančia hiperaktyvaus HRAS baltymo, turinčio įtakos ląstelių augimui ir dalijimuisi, sintezę. Su Costello sindromu audiniuose vyksta nuolatinis aktyvus ląstelių dalijimasis, prisidedantis prie gerybinių ir piktybinių navikų formavimosi.
Liga paveldima autosominiu recesyviniu būdu. Iki šiol didelė dalis
„Ros Vestn Perinatol Pediat 2010“; 5: 27-30
Korespondencijos adresas: Vasina Tamara Nikolaevna - medicinos mokslų kandidatė, docentė, vadovė. skyrius vaikų ligos su vaikų chirurgijos kursu Oryol medicinos institute
Zubtsova Tatjana Ivanovna - medicinos mokslų kandidatė, doc. skyriai 302028 Oryol, g. Oktyabrskaya, 4
Stavtseva Svetlana Nikolaevna - Oryolio perinatalinio centro genetikė
Kirsanova Tatjana Aleksandrovna - Oryol Perinatalinio centro ultragarsinės diagnostikos gydytoja 302019 Oryol, st. Generolas Zhadovas, 4
dovs turi spontaniškas genų mutacijas, o brolių ir seserų sindromo nustatymo galimybė yra nedidelė. Nepaisant to, yra atvejų, kai probando broliai ir seserys pasikartoja. Prenatalinė diagnozė įmanoma, jei šeimos nariai turi HRAS geno mutaciją. Dauguma pacientų, sergančių Costello sindromu, yra nevaisingi.
Sindromo diagnozė nustatoma remiantis būdingais klinikiniais simptomais ir molekulinių genetinių tyrimų rezultatais. Kruopštus prenatalinės istorijos tyrimas gali suteikti šią informaciją. Atlikus ultragarsinį tyrimą, vaisiui pasireiškė brachicefalija, galima aptikti žastikaulio ir šlaunikaulio sutrumpėjimą, 90% atvejų yra polihidramnionai. Aprašomos įvairios vaisiaus prieširdžių tachikardijos formos. Tačiau dauguma vaisiaus fenotipo ypatybių nėra specifinės, todėl prenatalinės diagnozės klausimas paprastai nekyla.
Formalūs Costello sindromo diagnostikos kriterijai dar nėra sukurti, tačiau žinomi pagrindiniai, unikalūs ligos simptomai, dėl kurių pacientai atpažįstami bet kuriame amžiuje. Naujagimio periodu atkreipiamas dėmesys į santykinę makrocefaliją, būdingą veidą, turintį didelę burną, storomis lūpomis, per didelį odos lankstymą, platų nosies tiltą, didelę kaktą ir epicantą. Pats įspūdingiausias klinikinis
simptomas neabejotinai yra disfagija (95% vaikų), kuri nuo gimimo sukelia didžiules maitinimo problemas, todėl labai sumažėja fizinio vystymosi tempai. Bandant žįsti kūdikį ar per spenelį, jis patiria aštrų nerimą („streso būsena“), rago atsisakymą. Tuo pat metu išsaugomas apetitas ir čiulpimo refleksas. Maitinimo problemos tampa tokios rimtos, kad daugeliu atvejų joms reikia atlikti nazogastrinį vamzdelį, atlikti gastrostomiją ar atlikti „Nielsen“ lėšų kaupimą. Tokiu atveju vaikai gali likti gastrostomos nešiotojais iki 4–6 metų.
Tarp bendrų požymių verta paminėti gilų odos sulankstymą ant delnų ir pėdų, delnų-plantacijų hiperkeratozę, odos hiperpigmentaciją natūraliose raukšlėse išilgai pilvo vidurio, hiperpigmentaciją spenelių areoloje. Dažnai yra papilomos aplink burną, nosies ir perianalio vestibiulyje. 50% atvejų nustatomos įvairios išvaržos. Atkreiptinas dėmesys į sunkią hipotenziją, staigų fizinio, motorinio ir neuropsichinio vystymosi uždelsimą, nerimą, dirglumą.
Jau pirmaisiais gyvenimo metais gali atsirasti plaučių arterijos vožtuvo stenozės požymiai (juos galima nustatyti nuo gimimo), hipertrofinė kardiomiopatija, supraventrikulinė tachikardija. Gali susidaryti hidrocefalija, aprašyti epilepsijos priepuoliai.
4–12 metų vaikai, išlaikydami būdingus fenotipinius požymius, turi trumpą ūgį ir ortopedines problemas (kyphoscoliosis, torticollis, hipermobilumas sąnariuose). Tokiame amžiuje jie pradeda savarankiškai valgyti tiek tirštą, tiek skystą maistą, atsikratę gastrostomos. Jie išsiskiria socialumu, draugiškumu ir turi tam tikrą humoro jausmą. Tačiau intelekto trūkumas išryškėja.
Paaugliams, sergantiems „Costello“ sindromu, būdingi klasikiniai veido bruožai, garbanoti plaukai, nosies fibromatozė, krūtinės papilomos, hiperkeratozė, hiperpigmentacija, trumpas ūgis, ortopediniai sutrikimai („aptempta kulno sausgyslė“ ir pėdų deformacija), protinis atsilikimas. Yra brendimo vėlavimas ar sutrikimas. Dėl augančios kyphoscoliosis, plonėjantys plaukai ir sensta oda, pacientai atrodo vyresni nei jų amžius. Piktybinių navikų (rabdosarkomos, neuroblastomos) rizika yra didelė.
Diferencinė Costello sindromo diagnozė kūdikystėje atliekama esant kardiofascio-odos sindromui (CFC), Noonan, Beckwith-Wiedemann, Simpson-Galabi, Williams sindromams. Tačiau, pasak ekspertų, fenotipas
kostello sindromo negalima supainioti su jokiu kitu žinomu genų sutrikimu. HRAS geno mutacijos nustatymas molekuliniais genetiniais metodais patvirtina diagnozę.
Ligos apraiškų gydymas nuo naujagimio laikotarpio yra susijęs su tinkamos mitybos užtikrinimu, kuriam dažniausiai reikia naudoti nazogastrinį vamzdelį ir įdiegti gastrostomiją. Dėl gastroezofaginio refliukso ligos išsivystymo dažnai reikia atlikti „Nielsen“ linksmąjį doplikacijos operaciją. Net ir pasiekus amžiui reikalingą kalorijų kiekį, augimo sulėtėjimas išlieka. Vaikus, turinčius širdies problemų, stebi ir gydo kardiologas pagal turimus standartus. Skeleto deformacijai reikia medicininės fiksacijos, fizinės terapijos, o kartais ir operacijos. Pažintinės funkcijos pastebimai pagerėja dėl anksti pradėtų individualių ugdymosi elgesio programų. Stebint vyresnio amžiaus pacientus, gali būti nustatyti hipoglikemijos atvejai. Tokiems vaikams reikalinga endokrinologo priežiūra. Internete yra svetainė, skirta susisiekti su šeimomis, turinčiomis vaikų, sergančių Costello sindromu.
Pateikiame savo klinikinės diagnozės ir vaiko, turinčio Costello sindromą, stebėjimo atvejį.
Mergaitė R., paguldyta į 1 mėnesio vaikų regioninės ligoninės Orelio somatinį skyrių dėl kūno svorio praradimo, išmatų susilaikymo, vaiko nerimo, sunkumų maitinant.
Gyvenimo ir ligos anamnezė: motina yra 30 metų, laiko save ir vaiko tėvą sveiką. Paveldimumą sunkina motinos senelės cukrinis diabetas. 1-asis nėštumas, pageidaujamas. Ultragarsinis tyrimas nustatė 13–14 nėštumo savaitę atliekant 7,3 x 5,5 mm kaklo aneksinę masę, nesusijusią su stuburu ir kauline kaukole (kaklo cista ?, nesubrendusi teratoma?). Be to, buvo nustatytas apykaklės erdvės plėtimasis iki 2,7 mm. Tiriant 14–15 nėštumo savaitę, dešinėje pusėje buvo nustatyta ir 5,2x5,0 mm dydžio neapčiuopiama kaklo masė. Po pakartotinių tyrimų, kuriuose dalyvavo genetikas Orelio mieste, moteris buvo nusiųsta į Akušerijos ir ginekologijos tyrimų institutą (Maskva) diagnozuoti įtariamą vaisiaus kaklo cistinę higromą. Prenatalinės vaisiaus patologijos serumo žymenų tyrimas, atliktas 16 nėštumo savaitės, parodė, kad α-fetoproteino lygis sumažėjo iki 0,43 Mot. Buvo nutarta pratęsti nėštumą.
19 nėštumo savaitę buvo pasiūlyta, kad yra žiaunios vaisiaus sinusai, kurie vėliau nebuvo vizualizuoti. Ultragarsinis tyrimas 23–24 nėštumo savaitę parodė polihidramnionus, rodančius sumažėjusį rijimą
Vasina T.N. et al. Klinikinė retos paveldimos ligos diagnozė - Costello sindromas
vaisiaus veikla. Nuo 24 savaičių nėštumas prasidėjo su gimdos kaklelio nepakankamumo požymiais, placentos hiperplazija; 29, 31, 37 nėštumo savaitę moteris buvo gydoma ligoninėje dėl pielonefrito.
Pirmieji gimimai 38–39 nėštumo savaitę. Pirmojo laikotarpio trukmė yra 9 valandos; II laikotarpis - 20 minučių, bevandenis intervalas - 4 valandos 50 minučių. Buvo pastebėti polihidramnionai, amniono skystis buvo žalias, užpakalinis skystis - rudai žalias. Dėl amniono skysčio išsiurbimo viršutiniai kvėpavimo takai buvo dezinfekuoti, 13 minučių atlikta dirbtinė plaučių ventiliacija. Apgaras surinko 7/8 balus, gimimo svoris 2980 g, ilgis 49 cm, galvos apimtis 34 cm. Motinystės ligoninėje buvo pastebėti CNS depresijos sindromo požymiai ir daugybinė disembryogenezės stigma, atlikta infuzija ir patogenezinis gydymas.
5 gyvenimo dieną mergaitė buvo perkelta į vaikų regioninės ligoninės Orelio naujagimių patologijos skyrių, kur 2 savaites buvo gydoma diagnozuojant perinatalinį hipoksinį smegenų pažeidimą, II laipsnio smegenų išemiją, depresijos sindromą; intrauterininis hipotrofinio augimo sulėtėjimas; daugialypės disembriogenezės stigmos. Vaikas turėjo ryškų hipersoft veido, kaklo odą; gilus sulenkimas ir kryžminis raištis ant kojų; hipertelorizmas delno sumušimų; Didelė burna; platus nosies tiltas; ausų dismorfizmas; galvos odos ir kaklo galo hemangiomos; trumpas kaklas (žr. paveikslėlį). Ją konsultavo genetikas; buvo įtariamas Šere-Shevsky-Turner sindromas. Atliktas kariotipas atskleidė normalų 46, XX kariotipą. Papildomas ultragarsinis tyrimas atskleidė hipoksinio-išeminio smegenų pažeidimo požymius (2,6 mm skersmens šoniniai skilveliai); pyeloectasia kairėje 6,3 mm.
Gautas gydymas: šėrimas „Nan“, vitamino B6, raumenų ATP, kavintono, espu-mizano, geriamojo bifidumbakterino mišiniu, masažas. Vaikas čiulpė vangiai, bet nesigailėjo, išmatos buvo taisyklingos. 2920 g kūno svorio ji buvo išleista prižiūrint vietos pediatrui ir neuropatologui ambulatoriškai.
Namuose motina, maitindama kūdikį, susidūrė su rimtomis problemomis: po kelių čiulpimo judesių mergaitė pradėjo smarkiai jaudintis, nusisuko nuo spenelio ir priešinosi maitinimo atnaujinimui. Po kurio laiko ji trumpam užmigo, tada atsibudo, vėl pradėjo godiai čiulpti ir lygiai taip pat staiga nustojo maitintis. Pamažu dingo nepriklausoma kėdė. 1 mėnesio metu pasimatyme su pediatru poliklinikoje kūno svoris buvo 2700 g., Diagnozavus III laipsnio netinkamą mitybą, vaikas buvo išsiųstas į vaikų zoną.
Paveikslėlis. Naujagimis su Costello sindromu.
nauja ligoninė.
Priėmimo būklė buvo sunki dėl sunkios hipotrofijos. Tyrimo metu jis neigiamas, šauksmas garsus, menkas. Ramina su žinduku, energingai čiulpia. Iš buteliuko mišinys ir vanduo pradeda aktyviai čiulpti, bet ne daugiau kaip 10 ml, tada kyla aštrus nerimas, verkimas, išvengia spenelio. Regurgitacija ir vėmimas nebuvo pastebėti. Stebint vaiką, disfagijos įspūdis sustiprėjo.
Mergaitė turėjo savitus fenotipinius bruožus: ji buvo neproporcinga dėl gana didelio galvos dydžio (apimtis 36,5 cm), jos kūnas ir galūnės buvo plonos. Oda yra sausa, gaiviai blyški, ant veido ir kaklo yra per daug raukšlių; gilios odos raukšlės ant delnų ir padų. Hiperpigmentuojami spenelių ploteliai, pilvo vidurio linija, virkštelės žiedas, natūralios raukšlės. Yra bambos išvarža ir vidurio pilvo išvarža. Akies plyšiai su epicanthus ir hipertelorizmu, kartais supanašėjantis strabismas. Platus nosies tiltas, aukštas „gotikinis“ gomurys, kapiliaras
nosies vestibiulės hemangioma, nasolabialinis trikampis. Auksinės yra meiliai susuktos, žemai nusileidusios. Didelis 2 x 2 šrifto fontanas, šiek tiek krinta. Kvėpuoti berniukas, nėra dusulio. Širdies garsai garsūs, nėra triukšmo, pulsas 140 per minutę. Pilvas yra minkštas, kepenys ir blužnis yra palpuojami pakrantės arkos krašte. Išmatos po klizmos yra raumeningos, negausios. Lytiniai organai formuojami pagal moterišką tipą. Esant neurologinei būklei: pusiau sulenkta laikysena, vidutinio sunkumo spontaninis motorinis aktyvumas, rankų drebėjimas, galūnių raumenų distonija.
Atliekant bendrąjį ir biocheminį kraujo tyrimą, patologinių pokyčių nepastebėta. Pagal ultragarsinį tyrimą kepenys, blužnis, skrandis, antinksčiai nepakinta; kairioji pyelektazė 7,8 mm. Neurosonografija: šoniniai 3,5 mm skilveliai, 2 mm tarpasferinis įtrūkimas, smegenyse patologinių pokyčių nenustatyta. Okulistų patologija nebuvo nustatyta. ENT gydytojas pastebėjo anatomiškai trumpą kaklą, siaurą nosiaryklę, aukštą epiglotito padėtį. Echokardiografijoje nebuvo jokių anatominių defektų. Pasitarus su chirurgu, siekiant išsiaiškinti disfagijos priežastis ir pašalinti įgimtus virškinimo trakto apsigimimus, buvo atlikta ezofagogastroduodenoskopija, kuri neatskleidė jokios organinės patologijos. Stemplės, skrandžio, žarnyno kontrastinė fluoroskopija: kardiospazmo, pylorospasm nuotrauka; sulėtėja evakacija iš skrandžio. Gydymas buvo atliktas: maitinimas Alfare mišiniu po 3 valandų, naudojant nazogastrinį vamzdelį, espumisaną, motiliumą, el.
automobilis, kreonas, bifidumbakterinas.
Vaiką iš naujo konsultavo genetikas, remiantis klinikiniais duomenimis, buvo pasiūlyta, kad yra Costello sindromas. Buvo rekomenduota konsultacija ir galimas tyrimas Rusijos medicinos mokslų akademijos Medicininių genetinių tyrimų centre. Mergaitė buvo išrašyta pagerėjus jos būklei, prižiūrint vietiniam pediatrui, neuropatologui ir genetikui, su rekomendacijomis, kaip maitinti mėgintuvėlius mišiniais, kurių pagrindas yra baltymų hidrolizatai, ir energgotropinę terapiją. Tolesnis dinamiškas vaiko stebėjimas leidžia nustatyti spėjamą diagnozę, nes yra pagrindiniai ligos diagnostiniai požymiai:
Disfagija;
Fizinio vystymosi tempo vėlavimas;
Gilus psichomotorinio vystymosi atsilikimas;
Hiper kaktos sulankstymas;
Gilios odos raukšlės ant delnų ir pėdų;
Virkštelės žiedo hiperpigmentacija;
Nosies vestibulo hemangioma;
Būdingi veido bruožai (hipertelorizmas akių plyšiuose, storos, įspaustos lūpos, didelė burna, liežuvis, platus nosies tiltelis);
Atsirandantis hidrocefalinis sindromas.
Šalies literatūroje nepavyko rasti ataskaitų apie pacientų, sergančių Costello sindromu, stebėjimus. Pateiktas retos genetinės ligos klinikinės diagnozės atvejis yra unikalus ir turi mokslinį bei praktinį susidomėjimą įvairių specialybių gydytojams.
LITERATŪRA
1. Gripp K.W., Lin A.E., Stabley D.L. et al. HRAS mutacijų analizė Costello sindromo metu: genotipo ir fenotipo koreliacija // Am. J. Med. Genetas. 2006. tomas 140A. 1-7 psl.
2. Kozlova S.I., Demikova N.S., Semanova E, Blinnikova O.E. Paveldimi sindromai ir medicininės genetinės konsultacijos. M .: Praktika, 1996. S. 122–123.
3. Gripp K. W., Lin A. E., Nicholson L. ir kt. Tolesnis fenotipo, gauto iš BRAForMEKI gemalo linijos, apibrėžimas
mutacija padeda atskirti kardio-facio-cutaneos sindromą nuo Costello sindromo // Am. Y. Med. Genetas. 2007. Vol. 143A. P. 1472–1480.
4. Kerr B, Delrue M.-A., Sigaudy S. ir kt. Genotipo ir fenotipo koreliacija Costello sindromu: HRAS mutacijų analizė 43 atvejais // J. Med. Genetas. 2006. tomas 43. P. 401–405.
- Yoon S, Seger R. Tarpląsteliniu signalu reguliuojama kinazė: keli substratai reguliuoja įvairias ląstelių funkcijas. Augimo veiksniai. 2006; 24 (1): 21–44. doi: 10.1080 / 02699050500284218
- Rauen KA. RASopatijos. Metinė genomikos ir žmogaus genetikos apžvalga. 2013; 14 (1): 355-369. doi: 10.1146 / annurev-genom-091212-153523
- Bosas JL. RAS onkogenai žmogaus vėžyje: apžvalga. Vėžio tyrimai. 1989 m. Rugsėjo 1 d., 49 (17): 4682–4689.
- Carragher L, Pritchard C, Aldridge V, Giblett S, Jin H, Foster C ir kt. Pelių modeliai, skirti BRAF sukeltiems vėžiams. Biocheminių visuomenės sandoriai. 2007; 35 (5): 1329. doi: 10.1042 / bst0351329
- Wallace M, Marchuk D, Andersen L, Letcher R, Odeh H, Saulino A ir kt. 1 tipo neurofibromatozės genas: identifikuotas didelis transkriptas, pažeistas trims NF1 sergantiems pacientams. Mokslas. 1990; 249 (4965): 181–186. doi: 10.1126 / mokslas.2134734
- „Cizmarova M“, „Kostalova L“, „Pribilincova Z“, „Lasabova Z“, „Hlavata A“, „Kovacs L“ ir kt. Rasopatijos - trumpo ūgio dismorfiniai sindromai ir piktybinio naviko rizika. Endokrininės sistemos. 2013; 47 (04): 217–222. doi: 10.4149 / endo_2013_04_217
- „Kratz CP“, „Rapisuwon S“, „Reed H“, „Hasle H“, „Rosenberg PS“. Vėžys Noonan, Costello, kardiofaciokutaniniai ir LEOPARD sindromai. Amerikos medicinos genetikos žurnalas, C dalis: Medicininės genetikos seminarai. 2011; 157 (2): 83–89. doi: 10.1002 / ajmg.c.30300
- „Tartaglia M“, „Gelb BD“, „Zenker M“. Noonano sindromas ir kliniškai susiję sutrikimai. Geriausia praktika ir tyrimai Klinikinė endokrinologija ir metabolizmas. 2011; 25 (1): 161–179. doi: 10.1016 / j.beem.2010.09.002
- Malaquias AC, Brasil AS, Pereira AC, Arnhold IJP, Mendonca BB, Bertola DR ir kt. Augimo standartai pacientams, sergantiems Noonan ir panašiais sindromais su mutacijomis RAS / MAPK kelyje. Amerikos medicinos medicinos genetikos žurnalas, A dalis, 2012; 158A (11): 2700–2706. doi: 10.1002 / ajmg.a.35519
- Noordam K, van der Bürgt I, „Brunner HG“, Otten BJ. Ryšys tarp Noonano sindromo klinikinio sunkumo ir augimo, augimo hormonų (GH) sekrecijos ir atsako į GH gydymą. Vaikų endokrinologijos ir metabolizmo žurnalas, 2002; 15 (2). Doi: 10.1515 / jpem.2002.15.2.175.
- „Binder G“, „Neuer K“, „Ranke MB“, „Wittekindt NE“. PTPN11Mutacijos yra susijusios su nedideliu augimo hormonų atsparumu asmenims, sergantiems Noonano sindromu. Klinikinės endokrinologijos ir metabolizmo žurnalas. 2005; 90 (9): 5377-5381. doi: 10.1210 / jc.2005-0995
- van der Burgtas I Retųjų ligų žurnalas „Orphanet“. 2007; 2 (1): 4. doi: 10.1186 / 1750-1172-2-4
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H ir kt. PTPN11 mutacijos, koduojančios baltymo tirozino fosfatazės SHP-2, sukelia Noonano sindromą. Gamtos genetika. 2001; 29 (4): 465-468. doi: 10.1038 / ng772
- Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A ir kt. Funkcijos padidėjimas SOS1 mutacijos sukelia savitą Noonano sindromo formą. Gamtos genetika. 2006; 39 (1): 75–79. doi: 10.1038 / ng1939
- Zenker M, Horn D, Wieczorek D, Allanson J, Pauli S, van der Burgt I ir kt. SOS1 yra antras dažniausiai pasitaikantis Noonan genas, tačiau jis neturi reikšmingo vaidmens širdies ir veido odos srityje. Medicininės genetikos žurnalas. 2007; 44 (10): 651–656. doi: 10.1136 / jmg.2007.051276
- Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S ir kt. Funkcionalios RAF1 mutacijos sukelia Noonan ir LEOPARD sindromus su hipertrofine kardiomiopatija. Gamtos genetika. 2007; 39 (8): 1007-1012. doi: 10.1038 / ng2073
- Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R ir kt. Vakcinių ląstelių funkcijų padidėjimas dėl RAF1 sukelia Noonano sindromą. Gamtos genetika. 2007; 39 (8): 1013-1017. doi: 10.1038 / ng2078
- Gremer L, Merbitz-Zahradnik T, Dvorsky R, Cirstea IC, Kratz CP, Zenker M ir kt. Ląstelės KRAS mutacijos sukelia abejotinas biochemines ir fizines savybes, sukeliančias vystymosi sutrikimus. Žmogaus mutacija. 2011; 32 (1): 33–43. doi: 10.1002 / humu.21377
- Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D ir kt. Ribotas NRAS mutacijų spektras sukelia Noonano sindromą. Gamtos genetika. 2009; 42 (1): 27–29. doi: 10.1038 / ng.497
- Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F ir kt. GermlineBRAFmutacijos Noonan, LEOPARD ir kardiofaciocutaniniuose sindromuose: molekulinė įvairovė ir susijęs fenotipinis spektras. Žmogaus mutacija. 2009; 30 (4): 695–702. doi: 10.1002 / humu.20955
- Komatsuzaki S, Aoki Y, Niihori T, Okamoto N, Hennekam RCM, Hopman S ir kt. SHOC2 geno mutacijų analizė esant Noonan tipo sindromui ir esant hematologiniams piktybiniams navikams. Žmogaus genetikos žurnalas. 2010; 55 (12): 801–809. doi: 10.1038 / 2010.01.16
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma "Ayan A, Sarkozy A, Fodale V ir kt. SHOC2 mutacija skatina abejotiną baltymo N-miristoilinimą ir sukelia į Noonan panašų sindromą su laisvais anageno plaukais. Gamtos genetika. 2009; 41 (9): 1022-1026. doi: 10.1038 / ng.425
- Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, Lepri F ir kt. Heterozigotinės lytinių ląstelių mutacijos CBL naviką slopinančiame gene sukelia į Noonano sindromą panašų fenotipą. Amerikos žmogaus genetikos žurnalas. 2010; 87 (2): 250–257. doi: 10.1016 / j.ajhg.2010.06.015
- „Niemeyer CM“, „Kang MW“, „Shin DH“, „Furlan I“, „Erlacher M“, „Bunin NJ“ ir kt. Ląstelės CBL mutacijos sukelia vystymosi anomalijas ir yra linkusios į jaunatvinę mielomonocitinę leukemiją. Gamtos genetika. 2010; 42 (9): 794–800. doi: 10.1038 / ng.641
- Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K ir kt. RIT1 funkcinių funkcijų padidėjimas sukelia Noonan sindromą, RAS / MAPK kelio sindromą. Amerikos žmogaus genetikos žurnalas. 2013; 93 (1): 173–180. doi: 10.1016 / j.ajhg.2013.05.021
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Peržiūrėta 1 tipo neurofibromatozė. Pediatrija. 2009; 123 (1): 124–133. doi: 10.1542 / peds.2007-3204
- Sarkozy A, Digilio M, Dallapiccola B. Leopardo sindromas. Retųjų ligų žurnalas „Orphanet“. 2008; 3 (1): 13. doi: 10.1186 / 1750-1172-3-13
- Revencu N, Boon LM, Mendola A, Cordisco MR, Dubois J, Clapuyt P ir kt. „RASA1“ mutacijos ir susiję fenotipai 68 šeimose, sergančiose kapiliarų ir arteriovenine malformacija. Žmogaus mutacija. 2013; 34 (12): 1632-1641. doi: 10.1002 / humu.22431
- „Boon LM“, „Mulliken JB“, „Vikkula M.“ RASA1: kintamas fenotipas su kapiliarų ir arterijų ir venų apsigimimais. Dabartinė nuomonė genetikos ir plėtros srityje. 2005; 15 (3): 265–269. doi: 10.1016 / j.gde.2005.03.004
- Rauen KA. HRAS ir Costello sindromas. Klinikinė genetika. 2007; 71 (2): 101–108. doi: 10.1111 / j.1399-0004.2007.00743.x
- Siegel DH, Mann JA, Krol AL, Rauen KA. Dermatologinis Costello sindromo fenotipas: Raso disreguliavimo pasekmės plėtojant. Britų žurnalas apie dermatologiją. 2012; 166 (3): 601–607. doi: 10.1111 / j.1365-2133.2011.10744.x
- Niihori T, Aoki Y, Narumi Y, Neri G, Cavé H, Verloes A ir kt. Germalinės KRAS ir BRAF mutacijos sergant širdies-fazio-odos sindromu. Gamtos genetika. 2006; 38 (3): 294–296. doi: 10.1038 / ng1749
- Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. Fosfatazės holoenzimas, kurį sudaro Shoc2 / Sur8 ir katalitinis PP1 funkcijų vienetas, kaip M-Ras efektorius moduliuojant gegnių aktyvumą. Molekulinė ląstelė. 2006; 22 (2): 217–230. doi: 10.1016 / j.molcel.2006.03.027
- „Nava C“, „Hanna N“, „Michot C“, „Pereira S“, „Pouvreau N“, Niihori T ir kt. Širdies, veido ir odos bei Noonano sindromai, atsirandantys dėl RAS / MAPK signalizacijos kelio mutacijų: genotipo fenotipo ryšiai ir sutapimas su Costello sindromu. Medicininės genetikos žurnalas. 2007; 44 (12): 763-771. doi: 10.1136 / jmg.2007.050450
- Yoon G, Rosenberg J, Blaser S, Rauen KA. Kardiofazinio-odos sindromo neurologinės komplikacijos. Plėtros medicina ir vaikų neurologija. 2007; 49 (12): 894–899. doi: 10.1111 / j.1469-8749.2007.00894.x
- Bremsas H, Legiusas E. Legius sindromas, atnaujinimas, SPRED1 mutacijų molekulinė patologija. Keio medicinos žurnalas. 2013; 62 (4): 107–112. doi: 10.2302 / kjm.2013-0002-RE
- Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA ir kt. Vakcinių ląstelių funkcijų padidėjimo SOS1 mutacijos sukelia Noonan sindromą. Gamtos genetika. 2006; 39 (1): 70–74. doi: 10.1038 / ng1926
- Narumi Y, Aoki Y, Niihori T, Sakurai M, Cavé H, Verloes A ir kt. Klinikinės apraiškos pacientams, sergantiems SOS1 mutacijomis, svyruoja nuo Noonan sindromo iki CFC sindromo. Žmogaus genetikos žurnalas. 2008; 53 (9): 834-841. doi: 10.1007 / s10038-008-0320-0
- „Fabretto A“, „Kutsche K“, „Harmsen M-B“, „Demarini S“, „Gasparini P“, „Fertz MC“ ir kt. Du Noonan sindromo atvejai, kai yra sunkus kvėpavimo ir virškinimo trakto pažeidimas bei SOS1 mutacija F623I. Europos medicinos genetikos žurnalas. 2010; 53 (5): 322–324. doi: 10.1016 / j.ejmg.2010.07.011
- Wennerbergas K. „RAS superfamily“ iš pirmo žvilgsnio. Ląstelių mokslo žurnalas. 2005; 118 (5): 843-846. doi: 10.1242 / jcs.01660
- Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F ir kt. Kraniosinostozė pacientams, sergantiems Noonano sindromu, kurį sukelia lytinių takų KRAS mutacijos. Amerikos medicinos medicinos genetikos žurnalas, A dalis, 2009; 149A (5): 1036-1040. doi: 10.1002 / ajmg.a.32786
- „Stark Z“, „Gillessen-Kaesbach G“, „Ryan MM“, „Cirstea IC“, „Gremer L“, „Ahmadian MR“ ir kt. Dvi naujos gemalo KRAS mutacijos: išplėsti molekulinį ir klinikinį fenotipą. Klinikinė genetika. 2012; 81 (6): 590–594. doi: 10.1111 / j.1399-0004.2011.01754.x
- Oliveira JB, Bidere N, Niemela JE, Zheng L, Sakai K, Nix CP ir kt. NRAS mutacija sukelia žmogaus autoimuninį limfoproliferacinį sindromą. Nacionalinės mokslų akademijos leidiniai. 2007; 104 (21): 8953-8958. doi: 10.1073 / pnas.0702975104
- Kobayashi T, Aoki Y, Niihori T, Cavé H, Verloes A, Okamoto N ir kt. RAF1in Noonan sindromo ir susijusių sutrikimų molekulinė ir klinikinė analizė: serino 259 kaip svarbiausio mutantų aktyvavimo mechanizmo defosforilinimas. Žmogaus mutacija. 2010; 31 (3): 284–294. doi: 10.1002 / humu.21187
- Tidyman WE, Rauen KA. RASopatijos: Ras / MAPK kelio disreguliacijos vystymosi sindromai. Dabartinė nuomonė genetikos ir plėtros srityje. 2009; 19 (3): 230–236. doi: 10.1016 / j.gde.2009.04.001
- Swaminathan G, Tsygankov AY. Cbl šeimos baltymai: žiedo lyderiai reguliuodami ląstelių signalizaciją. Ląstelių fiziologijos žurnalas. 2006; 209 (1): 21–43. doi: 10.1002 / jcp.20694
- „Flynn DC“. Adapterio baltymai. Onkogenas. 2001; 20 (44): 6270-6272. doi: 10.1038 / sj.onc.1204769
- „Stowe IB“, „Mercado EL“, „Stowe TR“, „Bell EL“, „Oses-Prieto JA“, „Hernandez H“ ir kt. Žmogaus rasopatijos Legius sindromas ir neurofibromatozė-1 yra bendras molekulinis mechanizmas. Genai ir raida. 2012; 26 (13): 1421–1426. doi: 10.1101 / gad.190876.112
Kostello sindromas yra labai reta liga, pažeidžianti daugybę kūno sistemų, sukelianti trumpą ūgį, būdingus veido bruožus, augimą aplink nosį ir burną bei širdies problemas. Kostello sindromo priežastis nežinoma, nors įtariama genetinė mutacija. 2005 m. DePlavero (JAV) „DuPont“ vaikų ligoninės tyrėjai nustatė, kad genų mutacijos HRAS seka buvo 82,5% iš 40 žmonių, sergančių Costello sindromu.
Pasaulinėje medicinos literatūroje paskelbta tik apie 150 pranešimų apie „Costello“ sindromą, todėl neaišku, kaip dažnai šis sindromas iš tikrųjų pasireiškia ar kas yra labiau paveiktas.
simptomai
Būdingi „Costello“ sindromo simptomai:
- Sunkumas priaugti svorio ir augti po gimimo, dėl ko atsiranda trumpas ūgis
- Per daug laisva kaklo, delnų, kojų pirštų ir pėdų oda (cutis laxa)
- Nepiktybiniai navikai (papilomos) aplink burną ir šnerves
- Skiriamasis veido vaizdas, pavyzdžiui, didelė galva, žemai sudėti ausys su dideliais, storais žiedlapiais, storomis lūpomis ir (arba) plačiomis šnervėmis.
- Protinis atsilikimas
- Sutirštėjusi, sausa rankų ir kojų arba rankų ir kojų oda (hiperkeratozė)
- Nenormaliai lankstūs pirštų sąnariai.
Kai kuriems žmonėms gali būti ribojamas alkūnių judėjimas arba suspausta sausgyslė kulkšnies gale. Žmonėms, sergantiems „Costello“ sindromu, gali būti širdies defektų ar širdies ligų (kardiomiopatija). Yra didelis naviko augimo dažnis - tiek piktybinis, tiek nepiktybinis, taip pat susijęs su sindromu.
diagnostika
„Costello“ sindromo diagnozė nustatoma atsižvelgiant į vaiko, gimusio su sutrikimu, išvaizdą, taip pat kitus simptomus, kurie gali būti. Daugumai kūdikių, sergančių „Costello“ sindromu, sunku maitintis ir jie priauga svorio ir auga, todėl tai gali reikšti diagnozę. Ateityje diagnozei patvirtinti gali būti naudojami žinomų genų mutacijų, susijusių su Costello sindromu, genetiniai tyrimai.
gydymas
Specifinio „Costello“ sindromo gydymo būdo nėra, todėl medicininė priežiūra orientuojasi į esamus simptomus ir sutrikimus. Visiems žmonėms, sergantiems Costello sindromu, patariama atlikti širdies tyrimą, kad būtų patikrinta, ar nėra širdies defektų ir (arba) širdies ligų. Fizinė ir ergoterapija gali padėti asmeniui pasiekti savo vystymosi potencialą. Svarbus ilgalaikis naviko augimo, stuburo ar ortopedinių problemų, širdies ar kraujospūdžio pokyčių stebėjimas. Kostello sindromu sergančio žmogaus gyvenimo trukmei įtakos turės širdies sutrikimai ar vėžys, taigi, jei jie yra sveiki, sindromu sergantiems žmonėms gyvenimo trukmė gali būti normali.
Kostello sindromas yra retas genetinis sutrikimas, susijęs su uždelstu fiziniu ir psichiniu vystymusi.
Jis pažeidžia įvairias kūno dalis ir pasižymi laisvomis odos raukšlėmis, prastu raumenų tonusu ir kitomis problemomis.
Kitos komplikacijos yra piktybinių ir nepiktybinių navikų vystymasis, širdies ydos ir nenormalus širdies raumens augimas.
Dažnos širdies problemos apima hipertrofinę kardiomiopatiją, kuri yra širdies padidėjimas, silpninantis širdies raumenį, nenormalūs širdies plakimai ar aritmija ir kiti struktūriniai defektai.
Manoma, kad Costello sindromas, dar žinomas kaip fasiocutaneoskeletinis sindromas (FCS), paveikė 200–300 žmonių visame pasaulyje, tačiau daugiau atvejų gali būti nediagnozuota.
Greiti faktai apie „Costello“ sindromą
- Kostello sindromas yra labai retas ir juo serga 200–300 žmonių visame pasaulyje.
- Tai gali sukelti uždelstą vystymąsi, psichinę negalią, didelę galvą ir lūpas su žemomis ausimis bei palaidą odą.
- Kostello sindromas taip pat sukelia širdies problemas. Tai genetinė liga, paveikianti daugybę kūno sistemų.
- Neatliekamas gydymas ir nėra specialaus šios būklės gydymo būdo. Gydymu siekiama palengvinti įvairius sindromo aspektus, pavyzdžiui, hipertrofinę kardiomiopatiją ir specialųjį mokymąsi, skirtą mokymosi negaliai paremti.
simptomai
Daugelio „Costello“ sindromo požymių nėra gimus, tačiau jie atsiranda, kai kūdikis pradeda augti.
Gimimo svoris paprastai yra normalus arba šiek tiek didesnis nei vidutinis, tačiau kūdikis augs lėčiau nei dauguma kūdikių.
Simptomai yra šie:
- trumpas ūgis ir lėtas augimas
- Protinė negalia
- vystymosi vėlavimas
- sunku maitinti krūtimi
- didelė galva
- laisva oda, ypač ant rankų ir kojų
- gilūs raukšliai delnuose ir pėdų paduose
- žemos ausys, stori ausies raišteliai arba abu
- lanksčios jungtys
- didelė burna
- veido paviršius atrodo šiurkštus
- strabismas
- širdies problemos, įskaitant nenormalų širdies ritmą
- dantų problemos
- tanki achilo sausgyslė
- stori skauda ir nagai
komplikacijos
„Costello“ sindromas yra sudėtinga, daugelio sistemų būklė, galinti sukelti įvairių komplikacijų.
Kūdikiai negali maitinti žodžiu, kol jiems nėra 2–4 \u200b\u200bmetų arba maždaug tuo pačiu metu, kai jie pradeda kalbėti.
Širdies ir kraujagyslių problemos dažnai išryškėja nuo ankstyvos vaikystės, nors simptomai gali nepasireikšti vėliau. Tai apima įgimtus širdies defektus ir širdies hipertrofiją. Asmuo gali patirti tachikardiją ar greitą širdies plakimą, aritmiją ar nereguliarų širdies ritmą.
Makrocefalija arba smegenų pervargimas pastebėtas 50% pacientų. Vieno tyrimo duomenimis, tai gali sukelti Chiari apsigimimus - struktūrinį smegenų defektą, kurį rado 32 procentai žmonių. Priepuoliai pasireiškia nuo 20 iki 50 procentų šia liga sergančių žmonių.
Gali būti stebimas kaulų augimo sulėtėjimas, mažas kaulų tankis ir didesnė kaulų lūžių bei osteoporozės rizika. Vitamino D ir kalcio papildai gali padėti.
Navikai gali išsivystyti, dažniausiai papilomos, nedideli augliai, primenantys karpos, ypač aplink nosį, burną ir išangę.
Žmonės, sergantys „Costello“ sindromu, yra linkę vystytis tiek vėžiniams, tiek vėžiniams navikams. Vėžys apima rabdomiosarkomą, neuroblastomą ir laikiną ląstelių karcinomą.
Kitos dvi genetinės ligos, turinčios panašių simptomų, yra Noonano sindromas ir širdies ir kraujagyslių sindromas (CFC). Dėl sutampančių simptomų sunku diagnozuoti „Costello“ sindromą kūdikystėje.
priežastys
Kostello sindromas yra genetinis sutrikimas, atsirandantis dėl HRAS geno mutacijų. Tai yra genas, kuris nurodo kūnui gaminti baltymus, žinomus kaip H-Ras. H-Ras skatina ląstelių augimą ir dalijimąsi.
Dėl Costello sindromo atsirandančios HRAS geno mutacijos sukelia ląstelių augimą ir dalijimąsi visą laiką, o ne tik tada, kai joms liepiama tai padaryti.
Tai gali sukelti tiek vėžinį, tiek vėžinį naviko augimą ir greičiausiai kitus simptomus.
HRAS geno mutacija taip pat gali paveikti elastinių skaidulų gamybą audinyje. Šios skaidulos yra gyvybiškai svarbios tokioms struktūroms kaip plaučiai, oda ir stambiosios kraujagyslės, įskaitant aortą.
Pluoštas yra svarbus norint išlaikyti stiprius plaukus ir odą, užkirsti kelią ankstyvam seksui ir išlaikyti kraujagyslių bei plaučių audinio vientisumą ir stiprumą.
Kad išsivystytų Costello sindromas, turi būti paveldėtas tik vienas mutavusio HRAS geno egzempliorius. Šis genų paveldėjimo tipas yra žinomas kaip autosominis dominavimas.
Daugelis „Costello“ sindromo atvejų yra susiję su naujomis mutacijomis, kai nėra šeimos būklės anamnezės.
diagnostika
Kostello sindromas yra labai retas, todėl mažai tikėtina, kad gydytojas tuoj įtaria.
Gydytojas pirmiausia įvertins kūdikio ūgį, galvos dydį ir gimimo svorį.
Kitas etapas apima molekulinius genetinius tyrimus. Atliekama HRAS geno nuosekli analizė, siekiant nustatyti, ar nėra mutacijų, susijusių su Costello sindromu.
gydymas
Negalima išgydyti nei Costello sindromo, nei jokio specifinio gydymo, tačiau tokius sindromo aspektus kaip širdies liga, hipertrofinę kardiomiopatiją galima gydyti specialiai.
Būdai, kaip padėti asmeniui valdyti šią ligą:
- padėti kūdikiui įveikti maitinimo sunkumus kūdikystėje
- gydant širdies problemas
- specialiojo ugdymo teikimas
Tyrėjai ieško veiksmingo būdo, kaip gydyti ligą genetiniu lygmeniu.
Kita veikla apima:
- profesionali ir kineziterapija
- operacija pailginti achilo sausgyslę
- papilomų pašalinimas krioterapija
prognozė
Asmens, sergančio Costello sindromu, gyvenimo trukmė nebuvo oficialiai ištirta. Tyrimai rodo, kad jei įvyktų mirtingumas, tai gali būti komplikacijų, tokių kaip širdies problemos, rezultatas.